Title:
Take home message:
While accurate predictions for the 3D structures of many proteins from primary sequence are now within our grasp, understanding function from protein structure or sequence is far from solved. .虽然从一级序列中准确预测许多蛋白质的三维结构已经在我们的掌握之中,但从蛋白质结构或序列中理解功能还远远没有解决。
Main:
Introduction
Metagenomics: promises and perils
(1)Shotgun metagenomic sequencing的优点:Complete functional profile of an environment 环境的完整功能概况;Genomic context and taxonomy obtained through binning/assembly 通过汇编获得基因组背景和分类;Higher accuracy achievable with proximity-guided assembly and long-read sequencing methods 使用邻近引导装配和长读数测序方法可获得更高的准确度;Can be combined with other meta-omics analyses 可以与其他元组学分析相结合;Generally less biased than activity- and PCR-based methods 通常比基于活性和PCR的方法偏差更小
Shotgun metagenomic sequencing的缺点:High sequencing depth required to detect genes in low abundance 检测低丰度基因需要高测序深度;Computationally-intensive assembly and binning 计算密集型汇编;Challenging to infer function from sequence alone 仅从序列推断功能具有挑战性。
(2)Activity-guided screening的优点:Can lead to detection of new enzymes or folds catalyzing known reactions 可以导致检测催化已知反应的新酶或折叠;Well-developed methods to screen for industrially-relevant enzymes, e.g., lipases, cellulases 筛选工业相关酶的成熟方法,例如脂肪酶、纤维素酶;Inexpensive 便宜的;Activity-forward method guarantees enzymes are active and express well in E. coli 活性转移法保证了酶在大肠杆菌中的活性和良好表达
Activity-guided screening的缺点:Limited to genes and small to medium-sized gene clusters that are expressed in the screening host 限于在筛选宿主中表达的基因和小到中等大小的基因簇;Typically limited to types of reactions that can be screened rapidly通常限于可以快速筛选的反应类型;Can requires specific high-throughput screening equipment Can需要特定的高通量筛选设备;Can only screen for one type of reaction/function at a time 一次只能筛选一种类型的反应/功能
(3)PCR-based screening的优点:Sensitive for low-abundance sequences 对低丰度序列敏感;Detect variation within a single gene family at the level of single nucleotide changes 在单个核苷酸变化的水平上检测单个基因家族内的变异;Relatively inexpensive 相对便宜
PCR-based screening的缺点:• Requires conserved DNA motifs in target sequences 需要靶序列中保守的DNA基序;Not effective for detecting novel enzyme seqences or folds 对于检测新的酶序列或折叠无效;Little to no taxonomic information 很少或没有分类信息;PCR-bias against GC-rich sequences 针对富含GC序列的PCR偏差;hort reads make gene cluster context difficult to recover 短阅读使基因簇上下文难以恢复
(4)酶发现的几种范式
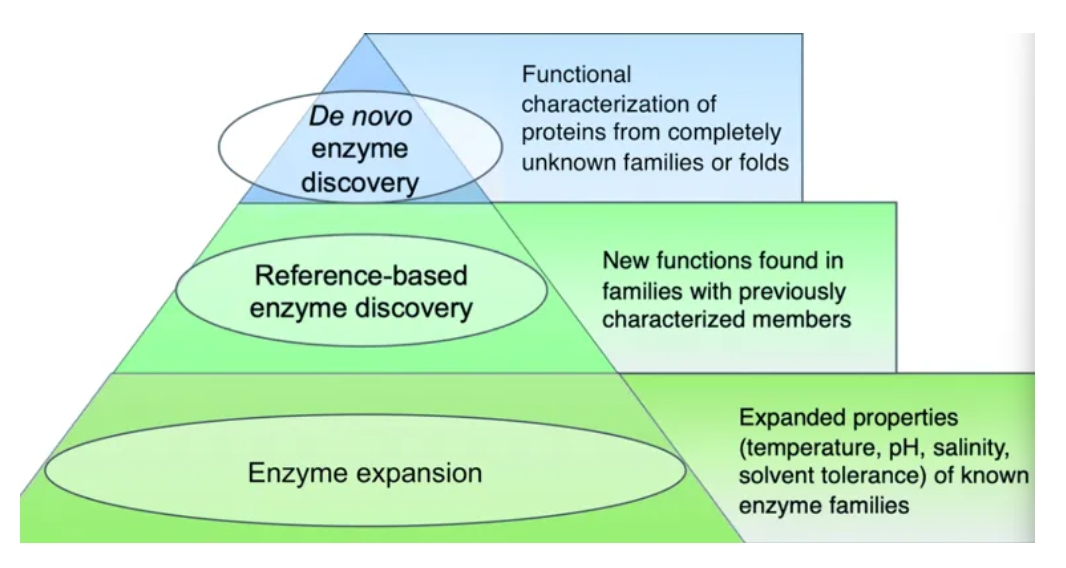
(5)What exactly is a ‘new’ enzyme? In this review, we conceptualize metagenomic enzyme discovery as a pyramid with three tiers (Fig. 1). The tip of the pyramid, which we refer to as de novo enzyme discovery, refers to the identification of entirely new types of biocatalysts. In other words, de novo enzymes must belong to protein folds or families without any functionally characterized members. 。“新”酶到底是什么?在这篇综述中,我们将宏基因组酶发现概念化为具有三层的金字塔(图1)。金字塔的顶端,我们称之为从头酶发现,指的是识别全新类型的生物催化剂。换句话说,从头酶必须属于没有任何功能特征成员的蛋白质折叠或家族。
(6)The second tier in the pyramid, which we call ‘reference-based enzyme discovery’, is the characterization of new reaction types within the context of already discovered protein families 金字塔的第二层,我们称之为“基于参考的酶发现”,是在已经发现的蛋白质家族的背景下表征新的反应类型。在基于参考文献的发现中,蛋白质折叠或家族中的一种或多种表征的酶是已知的,但是新发现的酶实际上功能不同。
(7)The base of the pyramid in Fig. 1, representing the largest fraction of metagenomic studies so far, refers to the discovery of enzymes with different substrate specificities or preferred reaction conditions including temperature, pH, salinity, or solvent preferences. Although often described as ‘enzyme discovery’ in the literature, we will refer to cases where the properties of a known enzyme class are extended as ‘enzyme expansion’ for clarity. 图1中金字塔的底部代表迄今为止宏基因组研究的最大部分,指的是具有不同底物特异性或优选反应条件(包括温度、pH、盐度或溶剂偏好)的酶的发现。尽管在文献中经常被描述为“酶的发现”,但为了清楚起见,我们将把已知酶类的性质扩展为“酶的扩展”。
Words:
NULL